I have published articles in journals including Physical Review B, The Journal of Physical Chemistry C, Chemical Communications, Physical Chemistry Chemical Physics (PCCP), the Journal of Physics: Condensed Matter, Solid State Communications, Solid State Ionics, the Journal of the American Ceramic Society and the Journal of Materials Chemistry A. In addition to original research articles, I have also published a high-profile Perspective article and my cover art was chosen for the PCCP outside front cover.

2020
Thematic Review
Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson, G. Ceder, Promises and Challenges of Next-Generation “Beyond Li-ion” Batteries for Electric Vehicles and Grid Decarbonization, Chemical Reviews, 121 (2021) 1623-1669..
 DOI: 10.1021/acs.chemrev.0c00767
DOI: 10.1021/acs.chemrev.0c00767
The tremendous improvement in performance and cost of lithium-ion batteries (LIBs) have made them the technology of choice for electrical energy storage. While established battery chemistries and cell architectures for Li-ion batteries achieve good power and energy density, LIBs are unlikely to meet all the performance, cost, and scaling targets required for energy storage, in particular, in large-scale applications such as electrified transportation and grids. The demand to further reduce cost and/or increase energy density, as well as the growing concern related to natural resource needs for Li-ion have accelerated the investigation of so-called “beyond Li-ion” technologies. In this review, we will discuss the recent achievements, challenges, and opportunities of four important “beyond Li-ion” technologies: Na-ion batteries, K-ion batteries, all-solid-state batteries, and multivalent batteries. The fundamental science behind the challenges, and potential solutions toward the goals of a low-cost and/or high-energy-density future, are discussed in detail for each technology. While it is unlikely that any given new technology will fully replace Li-ion in the near future, “beyond Li-ion” technologies should be thought of as opportunities for energy storage to grow into mid/large-scale applications.
2021
P. Hein, B.O.H. Grope, J. Koettgen, S. Grieshammer, M. Martin, Kinetic Monte Carlo simulations of ionic conductivity in oxygen ion conductors, Materials Chemistry and Physics, 257 (2021) 123767.
 PDF
PDF
 Supplement
DOI: 10.1016/j.matchemphys.2020.123767
Supplement
DOI: 10.1016/j.matchemphys.2020.123767
Ionic conductivities of solid-state materials are crucial for the performance of various applications ranging from batteries and fuel cells to resistive switching devices. The macroscopic ionic conductivity results directly from the microscopic energy landscape of ion diffusion. Lattice site energies and migration barriers depend on lattice defects such as vacancies and dopant ions in the local environment. The multiplicity of possible defect interactions with the migrating ion impedes the use of analytic models. While ab initio methods allow the calculation of the microscopic energy barriers for individual jumps, calculations of the macroscopic conductivity are computational very demanding, especially for more than 250 different materials and their possible ionic configurations as presented in this study. Kinetic Monte Carlo simulations allow the simulation of the ionic conductivity based on ab initio data and bridge the gap between microscopic jump events and the macroscopic conductivity. In this work, we discuss the Kinetic Monte Carlo method and its application to oxygen ion conductors for the example of doped ceria. We demonstrate how Kinetic Monte Carlo simulations can be accelerated to be 100 times faster with preserved high accuracy. Moreover, we report how the accuracy of Kinetic Monte Carlo simulations is improved with a large interaction radius and minimal computational expenses.
2020
J. Koettgen, C.J. Bartel, J. Shen, K.A. Persson, G. Ceder, First-principles study of CaB12H12 as a potential solid-state conductor for Ca, Physical Chemistry Chemical Physics 22 (2020) 27600-27604.
DOI: 10.1039/D0CP04500D
Calcium dodecahydro-closo-dodecaborate, CaB12H12, was calculated to have a percolating Ca migration path with low activation barrier (650 meV). The formation of Ca vacancies required for diffusion was calculated to be thermodynamically feasible by substitution of Ca with Al, Bi, or a number of trivalent rare-earth cations.
2020
HOT Article Communication
J. Koettgen, C.J. Bartel, G. Ceder, Computational investigation of chalcogenide spinel conductors for all-solid-state Mg batteries, Chemical Communications 56 (2020) 1952-1955.
PDF
Supplement
DOI: 10.1039/C9CC09510A
Seven MgLn2X4 (Ln = lanthanoid, X = S, Se) spinels are calculated with density functional theory to have low barriers for Mg migration (<380 meV) and are stable or nearly stable (within 50 meV per atom of stability with respect to competing structures and compositions). As the size of the Ln increases, Mg mobility is found to increase, but stability in the spinel structure is found to decrease.
2020
J. Koettgen, S. Grieshammer, G. Dück, G. Ulbrich, M. Lerch, M. Martin, Ab initio and experimental oxygen ion conductivities in Sm-Zr and Gd-Zr co-doped ceria, Solid State Ionics, 355 (2020) 115422.
PDF
Supplement
DOI: 10.1016/j.ssi.2020.115422
The oxygen ion conductivities of Sm doped ceria and Gd doped ceria are the highest for a rare-earth doped cerium oxide, which can be applied in solid oxide fuel cells (SOFC), high-temperature electrolysis with solid oxide electrolysis cells (SOEC), and high-temperature batteries. At the same time, zirconium oxide is a common impurity that has been shown to reduce the ionic conductivity in this type of doped ceria material. Doping with zirconia does not create additional oxygen vacancies and the conductivity is only influenced by the additional interactions between the Zr ions and either the rare-earth dopants or the oxygen vacancies. In this work, ceria co-doped with Sm-Zr and Gd-Zr is investigated using impedance spectroscopy experiments as well as density functional theory calculations and Kinetic Monte Carlo simulations. Co-doping Sm-doped and Gd-doped ceria with Zr decreases the conductivity significantly in experiments and simulations. The strong influence on the conductivity is due to the effective trapping of the oxygen vacancies by the zirconium ions.
2020
Communication
J. Koettgen, M. Martin, Infinite dilution in doped ceria and high activation energies, Solid State Communications, 314-315 (2020) 113939.
PDF
DOI: 10.1016/j.ssc.2020.113939
Oxygen ion diffusion determines the performance of materials in energy conversion, energy storage and catalysis. For nominally pure cerium oxide, experiments measure high activation enthalpies while calculations predict low activation enthalpies. Moreover, for doped oxides, e.g. doped ceria, experiments show a high activation enthalpy for both pure ceria and for high dopant fractions, leading to a minimum in activation enthalpy for small dopant fractions. While for high dopant fractions the increase in activation enthalpy is correlated with the association of oxygen vacancies and dopant ions, which are both created by doping, the minimum in activation enthalpy is assumed in the literature to be related to the maximum in ionic conductivity at similar dopant fractions. In this study, density functional theory (DFT) calculations and Kinetic Monte Carlo (KMC) simulations are combined in order to calculate the ionic conductivity and activation enthalpy in doped oxides. We show that the experimental ionic conductivity and activation energy in nominally pure cerium oxide is dominated by impurities. We resolve the discrepancy between activation enthalpies of nominally pure oxides in experiments as opposed to calculations. This will lead to a more comprehensive understanding of the oxygen ion conductivity and its underlying atomistic mechanisms. Moreover, such a focus will be of great benefit to the future development of sustainable and efficient materials.
2020
J. Koettgen, G. Dück, M. Martin, The oxygen ion conductivity of Lu doped ceria, Journal of Physics: Condensed Matter 32 (2020) 265402.
PDF
DOI: 10.1088/1361-648X/ab7d64
The oxygen ion conductivity of polycrystalline samples of Lu doped ceria is studied using impedance spectroscopy. Lutetium doped ceria is of particular interest as Lu has a similar ionic radius as the host cation Ce. The change of the ionic conductivity as a function of the Lu dopant fraction is investigated in detail revealing a similar behavior as Sm doped ceria that has one of the highest ionic conductivity in ternary cerium oxides. In comparison with simulations, the experimental dependence of the conductivity on the dopant fraction reveals that migration barriers for oxygen vacancy jumps around Lu ions are slightly higher than for jumps in pure ceria. The absolute conductivity is small due to the strong trapping of oxygen vacancies near Lu dopants.
2020
J. Koettgen, M. Martin, The ionic conductivity of Sm-doped ceria, Journal of the American Ceramic Society, 103 (2020) 3776-3787.
PDF
DOI: 10.1111/jace.17066
The oxygen ion conductivity of polycrystalline samples of Sm-doped ceria and of Gd-doped ceria is studied as a function of doping fraction and temperature using impedance spectroscopy allowing the separation of bulk and grain boundary conductivity. The introduction of a fine spacing for the Sm dopant fraction allows the clear identification of the dopant fraction leading to the largest bulk conductivity. At 267°C, the largest bulk conductivity is shown for Ce0.93Sm0.07O1.965. With increasing temperature, indications of an increase in the dopant fraction, which leads to the maximum in conductivity, are found. For the grain boundary conductivity, the maximum appears at larger dopant fractions compared to the bulk conductivity. The largest total conductivity for both dopants is again found for Sm-doped ceria. In literature, different syntheses and sample preparation methods led to larger total conductivities for Gd-doped ceria. In this work, we demonstrate that the variation of sintering conditions leads to scattering in the conductivity over one order of magnitude. Finally, we demonstrate that, in nominally pure cerium oxide, impurities dominate the ionic conductivity.

2019
J. Koettgen, M. Martin, The Effect of Jump Attempt Frequencies on the Ionic Conductivity of Doped Ceria, The Journal of Physical Chemistry C 123 (2019) 19437-19446.
PDF
DOI: 10.1021/acs.jpcc.9b06946
The macroscopic oxygen ion conductivity in doped ceria is determined by the microscopic activation energy barriers and jump attempt frequencies of oxygen ion jumps. While the influence of the local jump environment on the migration energy is widely investigated, its influence on the attempt frequency is rarely investigated. In this work, attempt frequencies in Sm, Yb, and Gd doped ceria are calculated using density functional theory. Moreover, ionic conductivities for varying local jump attempt frequencies in different jump environments are investigated using Kinetic Monte Carlo simulations. For doping along the migration pathway, where the migrating oxygen ion passes between two adjacent cations, large dopants lead to an increase and small dopants to a decrease in the attempt frequency. Sm doping in nearest neighborhood to the start position of the migrating oxygen vacancy also leads to an increase in attempt frequency. Kinetic Monte Carlo simulations show that at intermediate Sm dopant fractions oxygen vacancies frequently jump toward and away from dopants explaining why for Sm doped ceria one of the highest conductivities for a ternary cerium oxide was measured due to its low dopant-oxygen vacancy association in both nearest and next-nearest neighborhood.
2019
J. Koettgen, M. Martin, Coordination Numbers in Sm-Doped Ceria Using X-ray Absorption Spectroscopy, The Journal of Physical Chemistry C 123 (2019) 6333-6339.
PDF
DOI: 10.1021/acs.jpcc.8b10494
Sm-doped ceria has one of the highest ionic conductivities reported for rare earth-doped cerium oxides. The high oxygen-ion conductivity can be attributed to the creation of oxygen vacancies by doping and weak defect interactions between oxygen vacancies and dopants. Especially, oxygen vacancies in the nearest neighborhood to dopants decrease the conductivity due to trapping and blocking. In this work, the local structure around the Ce cations is investigated using the extended X-ray absorption fine structure. The resulting coordination numbers of cerium coordinated by oxygen are only marginally larger than in a random oxygen vacancy distribution, explaining the large ionic conductivity.
2018
HOT Article Perspective
J. Koettgen, S. Grieshammer, P. Hein, B.O.H. Grope, M. Nakayama, and M. Martin, Understanding the ionic conductivity maximum in doped ceria: trapping and blocking, Physical Chemistry Chemical Physics 20 (2018) 14291-14321.
PDF
Supplement
DOI: 10.1039/C7CP08535D

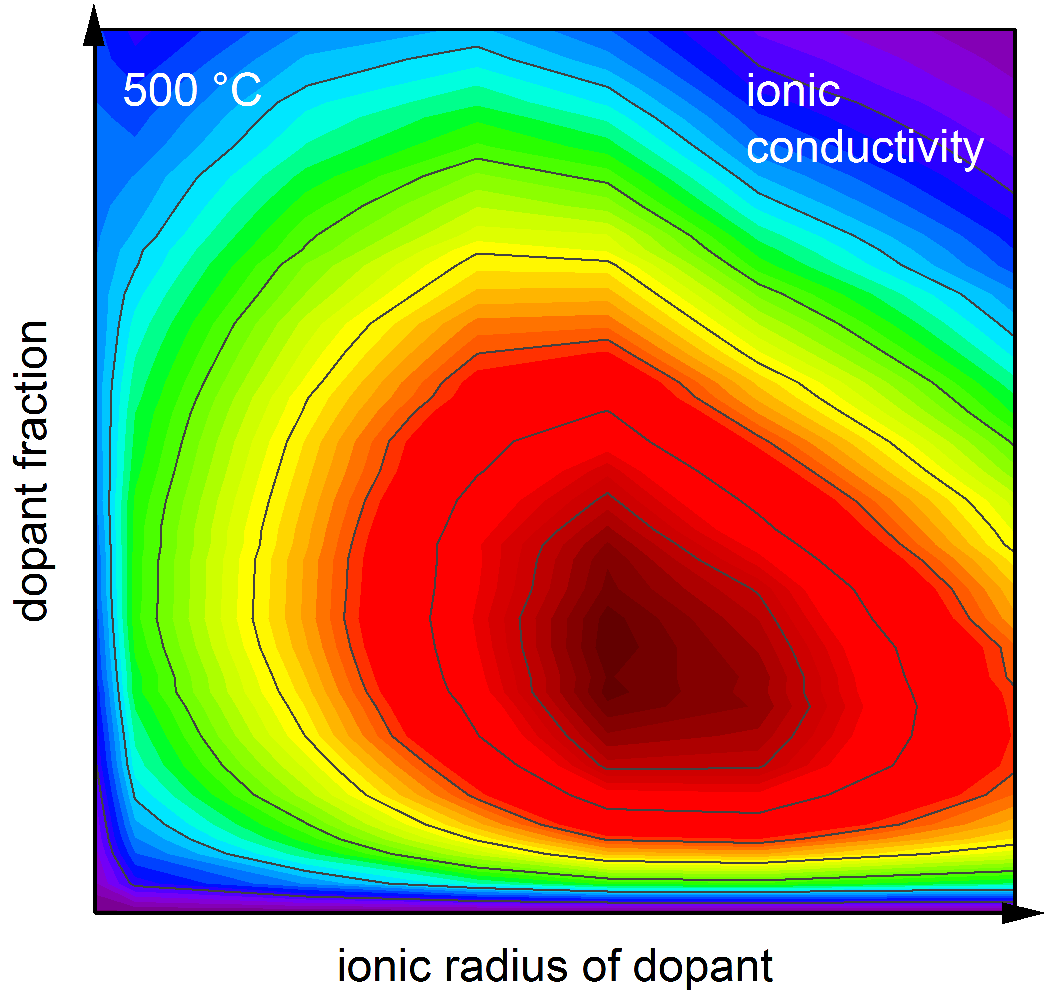
In our PCCP cover picture, we show how defects in solids influence the ionic conductivity. Doping oxides with lower valent dopant ions (blue spheres) creates oxygen vacancies (red cubes) that increase the ionic conductivity. Diffusing oxygen vacancies are caught by dopants. As dopants now hold the oxygen vacancies firmly, oxygen vacancies are immobilized. Further jumps of the oxygen vacancies are hindered and the ionic conductivity is reduced. This two‑step trapping process (catch‑and‑hold) varies with each dopant. The more frequently dopants catch and the longer they hold oxygen vacancies, the lower the ionic conductivity. The catch‑and‑hold principle of trapping thus determines which material has the highest ionic conductivity. Additionally, oxygen vacancies cannot escape through the gaps between tentacles of the dopants. Jumps of oxygen vacancies through these gaps are blocked by the tentacles. If two dopants are next to each other, oxygen vacancies that are caught rarely escape. The creation of oxygen vacancies by doping competes with the blocking of the tentacles of the dopants. Thus, blocking determines the concentration of dopants, leading to the highest ionic conductivity. In summary, our cover not only explains which fraction of dopant ions is the best. The picture also demonstrates which dopant leads to the highest possible ionic conductivity. This knowledge is crucial for the development of new materials for catalysis and energy conversion.
Materials with high oxygen ion conductivity and low electronic conductivity are required for electrolytes in solid oxide fuel cells (SOFC) and high-temperature electrolysis (SOEC). A potential candidate for the electrolytes, which separate oxidation and reduction processes, is rare-earth doped ceria. The prediction of the ionic conductivity of the electrolytes and a better understanding of the underlying atomistic mechanisms provide an important contribution to the future of sustainable and efficient energy conversion and storage. The central aim of this paper is the detailed investigation of the relationship between defect interactions at the microscopic level and the macroscopic oxygen ion conductivity in the bulk of doped ceria. By combining ab initio density functional theory (DFT) with Kinetic Monte Carlo (KMC) simulations, the oxygen ion conductivity is predicted as a function of the doping concentration. Migration barriers are analyzed for energy contributions, which are caused by the interactions of dopants and vacancies with the migrating oxygen vacancy. We clearly distinguish between energy contributions that are either uniform for forward and backward jumps or favor one migration direction over the reverse direction. If the presence of a dopant changes the migration energy identically for forward and backward jumps, the resulting energy contribution is referred to as blocking. If the change in migration energy due to doping is different for forward and backward jumps of a specific ionic configuration, the resulting energy contributions are referred to as trapping. The influence of both effects on the ionic conductivity is analyzed: blocking determines the dopant fraction where the ionic conductivity exhibits the maximum. Trapping limits the maximum ionic conductivity value. In this way, a deeper understanding of the underlying mechanisms determining the influence of dopants on the ionic conductivity is obtained and the ionic conductivity is predicted more accurately. The detailed results and insights obtained here for doped ceria can be generalized and applied to other ion conductors that are important for SOFCs and SOECs as well as solid state batteries.
2018
S. Grieshammer, S. Eisele, J. Koettgen, Modeling Oxygen Ion Migration in the CeO2-ZrO2-Y2O3 Solid Solution, The Journal of Physical Chemistry C 122 (2018) 18809-18817.
DOI: 10.1021/acs.jpcc.8b04361
Zirconium doping in cerium oxide, as a result of intentional material engineering or unintentional impurities, impacts material properties like reduction behavior and defect migration. In this study, we investigate the influence of zirconium doping on the conductivity of yttrium doped ceria using DFT+U calculations. We calculate the migration energies of oxygen ions for different jump environments containing yttrium and zirconium ions and compare the results to a simplified migration energy model. The small zirconium ions lead to strong distortions of the lattice, which results in deviation between calculated and modeled energies. Both the calculated and the modeled migration energies are used in kinetic Monte Carlo simulations to obtain the ionic conductivity for various dopant fractions. We identify three major influences on the ionic conductivity: the trapping of oxygen vacancies by dopant ions, the blocking effect, which alters the migration barriers around defects, and the lattice contraction due to zirconium doping.

2018
J. Koettgen, P. C. Schmidt, T. Bučko, and M. Martin, Ab initio calculation of the migration free energy of oxygen diffusion in pure and samarium-doped ceria, Physical Review B 97 (2018) 024305.
PDF
Supplement
DOI: 10.1103/PhysRevB.97.024305
In simple terms, you could describe us as physicists who filled a gymnasium with sand to measure the free energy activation barrier: Imagine that your high school coach asked you to jump over an obstacle, but he did not tell you whether or not to perform a somersault along the way. Probably, a decade ago, you would feel like oxygen ions jumping through a solid in a theoretical calculation. Back then, energy barriers for oxygen ion jumps were calculated from scratch. The resulting activation energy for diffusion corresponds to the height of the obstacle in your high school gymnasium. However, the level of entropy necessary to cross the activation barrier, i.e. jumping with or without a somersault, was ignored. Today, all vibrational modes at the top of the barrier can be investigated. In this article, we go one step further: We fill the gymnasium with sand until we can no longer see the obstacle. By filling up the double-well free energy potential for the oxygen ion jump, we see the activation energy and entropy of the migration barrier. The resulting migration free energy provides important catalytic and ion-conducting properties of ceria and this new standard method can be applied to a wide range of materials.
We have studied the free energy migration barriers ΔF‡ for oxygen diffusion in pure ceria and Sm-doped ceria for the temperatures 300, 700, and 1000 K. We used the density functional theory in the generalized gradient approximation and an additional Hubbard U parameter for the Ce 4f electronic states. We compare the results for the free energy deduced from three different methods. First, a static harmonic approach is applied in which the temperature dependent vibrational contributions to energy and entropy are deduced from the phonon frequencies of supercells with a fixed volume. Second, a static quasiharmonic approach is used in which a part of the anharmonicity effect is introduced via an implicit dependence of the harmonic frequencies on the thermally expanding cell volume. Third, the free energy barriers are calculated using metadynamics and molecular dynamics in which anharmonicity effects are naturally taken into account. The three methods examined in this study lead to distinctly different results. According to the harmonic approximation, the migration free energy difference ΔF‡ increases with increasing temperature due to an increasing entropic contribution. According to the quasiharmonic approximation, the migration free energy is independent of temperature. Finally, molecular dynamics predicts a thermally induced increase in the migration free energy. We conclude that temperature dependent experimental lattice constants cancel out the increasing entropic contribution with increasing temperature in the static quasiharmonic approach. The full consideration of anharmonicity effects in the metadynamics method again leads to a temperature dependent migration free energy.
2017
J.R. Köttgen, The relation between defect interactions, local structure and oxygen ion conductivity in the bulk of doped ceria, Dissertation, RWTH Aachen University, 2017.
PDF
DOI: 10.18154/RWTH-2017-01919
For the development of solid oxide fuel cells (SOFC) and high-temperature electrolysis an electrolyte, which has a high oxygen ion conductivity and a low electronic conductivity, is required. Potential candidates are fluorite-structured oxides such as doped zirconia (ZrO2) and doped ceria (CeO2). The latter allows a reduction in the operating temperature from 900 °C to 600 °C, and is, therefore, the main focus of this thesis. The central aim of this thesis is the detailed understanding of the relationship between defect interactions, the local structure (microscopic level) and the macroscopic oxygen ion conductivity in the bulk of fluorite-structured oxides. By combining ab initio density functional theory with Kinetic Monte Carlo simulations, both the local structure and the oxygen ion conductivity are predicted as a function of the doping concentration. These results are verified by accompanying experiments. In this way, a deeper understanding of the underlying mechanism is obtained in order to make better predictions about material properties. Therefore, this work provides a contribution to the study of sustainable and efficient energy storages.
2017
M. Martin, R.A. De Souza, J. Köttgen, F. Draber, and P. Hein, Defects and diffusion in solids: Application of new theoretical concepts, Bunsen-Magazin 19.3 (2017) 138-139.
2017
J. Koettgen, T. Zacherle, S. Grieshammer, and M. Martin, Ab initio calculation of the attempt frequency of oxygen diffusion
in pure and samarium doped ceria, Physical Chemistry
Chemical Physics 19 (2017) 9957-9973.
PDF
DOI: 10.1039/C6CP04802A
The rate of oxygen ion jumps in a solid oxide depends not only on the activation energy but also on the pre-exponential factor of diffusion. In order to allow a fully ab initio prediction of the oxygen ion conductivity in pure and samarium doped ceria, we calculated the attempt frequency for an oxygen ion jump from first principles combining DFT+U, the NEB method, phonon calculations and the transition state theory. Different definitions of the jump attempt frequency are presented. The equivalence of the Eyring and the Vineyard method is shown without restriction to the Gamma point. Convergence checks of the phonon mesh reveal that the common reduction to the Gamma point is not sufficient to calculate the attempt frequency. Calculations of Sm doped ceria revealed an increase of the prefactor. The attempt frequency for the constant pressure case in quasi-harmonic approximation is larger than the attempt frequency at constant volume in harmonic approximation. The calculated electronic energies, enthalpies and entropies of migration are in agreement with the experimental diffusion coefficients and activation energies.

2014
S. Grieshammer, B.O.H. Grope, J. Koettgen and M. Martin,
A combined DFT+U and Monte Carlo study on rare-earth
doped ceria, Physical Chemistry Chemical Physics 16
(2014) 9974-9986.
PDF
Supplement
DOI: 10.1039/C3CP54811B
In doped ceria, oxygen vacancies (cubes) and dopants (spheres) are the main defects. Oxygen vacancies repel each other. Dopants repel each other, but oxygen vacancies are attracted to dopants. The positions of the defects in thermal equilibrium can be determined using Metropolis Monte Carlo (MMC) simulations. The mobility and diffusion of the defects can be determined using Kinetic Monte Carlo (KMC) simulations. In both simulations, each move of a defect is rolled. In MMC, cubes and spheres act in a similar fashion to queens in chess. In KMC, all defects are kings.
We investigate the dopant distribution and its influence on the oxygen ion conductivity of ceria doped with rare earth oxides by combining density functional theory and Monte Carlo simulations. We calculate the association energies of dopant pairs, oxygen vacancy pairs and between dopant ions and oxygen vacancies by means of DFT + U including finite size corrections. The cation coordination numbers from ensuing Metropolis Monte Carlo simulations show remarkable agreement with experimental data. Combining Metropolis and Kinetic Monte Carlo simulations we find a distinct dependence of the ionic conductivity on the dopant distribution and predict long term degradation of electrolytes based on doped ceria.
2014
T. Leichtweiss, R.A. Henning, J. Koettgen, R.M. Schmidt, B.
Holländer, M. Martin, M. Wuttig, and J. Janek, Amorphous and
highly nonstoichiometric titania (TiOx) thin films close to
metal-like conductivity, Journal of Materials Chemistry A 2
(2014) 6631-6640.
PDF
DOI: 10.1039/C3TA14816E
Oxygen-deficient titanium oxide films (TiOx) have been prepared by pulsed laser deposition at room temperature. Samples in their as-deposited state have an average composition of TiO1.6, are optically absorbing and show electronic conductivities in the range of 10 S cm−1. The films are metastable and consist of grains of cubic titanium monoxide (γ-TiO) embedded in an amorphous TiO1.77 matrix. Upon annealing in an argon atmosphere the electrical conductivity of the films increases and comes close to metal-like conductivity (1000 S cm−1) at about 450 °C whereas the local structure is changed: nanocrystalline grains of metallic Ti are formed in the amorphous matrix due to an internal solid state disproportionation. The highly conductive state can be frozen by quenching. During heat treatment in an argon atmosphere a stoichiometric rutile TiO2 surface layer forms due to oxidation by residual oxygen. The combination of a highly conductive TiOx film with such an approximately 20 nm thick rutile cover layer leads to a surprisingly high efficiency for the water-splitting reaction without the application of an external potential.
2012
M. Lumeij, J. Koettgen, M. Gilleßen, T. Itoh, and R. Dronskowski, Detailed insights into the structural properties and oxygen-pathways in orthorhombic Ba0.5Sr0.5Co0.8Fe0.2O3–δ by electronic-structure theory, Solid State Ionics Vol. 222-223 (2012) 53-58.
PDF
DOI: 10.1016/j.ssi.2012.07.004
A number of structural properties of orthorhombic Ba0.5Sr0.5Co0.8Fe0.2O3–δ (BSCF) have been investigated by means of quantum-chemical calculations based on density-functional theory (DFT) and compared with experimental results. The role of the cation arrangements and the location of the oxygen vacancies within the orthorhombic structure have been evaluated and explained by means of bond-analytical techniques. Moreover, a detailed investigation of all oxygen pathways within orthorhombic BSCF has been performed, and the calculations show the existence of preferred oxygen pathways.
According to the publishers' licenses, be aware that the PDFs may not be further made available or distributed.