At Lawrence Berkeley National Laboratory and the University of California, Berkeley, I developed new cathode and electrolyte materials for Mg and Ca batteries. Ab initio density functional theory (DFT) calculations are used to determine the structure, thermodynamic stability, and Mg and Ca mobility of potential battery materials.
![]() DFT
MD
high-throughput computing
Materials Project
thermodynamic stability
materials design and discovery
DFT
MD
high-throughput computing
Materials Project
thermodynamic stability
materials design and discovery
My previous research at RWTH Aachen University focused on the relationship between defect interactions, local structure and oxygen ion conductivity in solids using theoretical and experimental methods. By combining ab initio density functional theory (DFT) with Kinetic Monte Carlo (KMC) simulations, both the local structure and the oxygen ion conductivity are predicted as a function of the doping concentration. These results are verified by accompanying experiments, for example extended X‑ray absorption fine structure (EXAFS) and impedance spectroscopy measurements.
![]() DFT
TST
phonon calculations
MD
metadynamics
KMC
MMC
impedance spectroscopy
XRD
EDX
SEM
XAS
XANES
EXAFS
DFT
TST
phonon calculations
MD
metadynamics
KMC
MMC
impedance spectroscopy
XRD
EDX
SEM
XAS
XANES
EXAFS
![]() DFT
high-throughput computing
Materials Project
thermodynamic stability
materials design and discovery
DFT
high-throughput computing
Materials Project
thermodynamic stability
materials design and discovery
High magnesium mobility at room temperature is required for solid-state electrolytes for all-solid-state Mg batteries. However, few materials provide a high magnesium mobility as the high charge density of magnesium leads to strong electrostatic interactions with the host lattice. We have identified seven magnesium chalcogenide spinels as possible conductors. MgLn2X4 (Ln = lanthanoid, X = S, Se) spinels are calculated with density functional theory to have low barriers for Mg migration. All materials are stable or nearly stable. Moreover, we discovered that the Mg mobility increases with larger lanthanoids, but the stability of the spinel structure decreases. Our findings demonstrate that Mg migration can be facilitated by placing Mg in the non-preferred tetrahedral coordination environment and increasing the interatomic distances. These materials design strategies to improve the Mg mobility are constrained by changes in the migration mechanism and the decreasing synthesizability of the material.

For the development of solid oxide fuel cells (SOFC) and high-temperature electrolysis with solid oxide electrolysis cells (SOEC), an electrolyte is required, which has a high oxygen ion conductivity and a low electronic conductivity. Potential candidates are fluorite-structured oxides such as doped zirconia (ZrO2) and doped ceria (CeO2). Doped ceria allows a reduction in the operating temperature from 900 °C to 600 °C, and is, therefore, the main focus of my research. In so doing, my work provides a contribution to the study of sustainable and efficient energy storages.
![]() DFT
KMC
impedance spectroscopy
XRD
DFT
KMC
impedance spectroscopy
XRD
For more than 50 years, it has been known that the ionic conductivity in doped oxides depends on the doping concentration, the choice of the dopant, and temperature. In fact, a maximum ionic conductivity exists. According to the literature, the ionic conductivity maximum appears when the increase in charge carrier concentration, caused by doping with a lower valent oxide, is compensated by the formation of associates of dopants with oxygen vacancies. The optimal dopant is assumed to be connected with a low level of associate formation. Beyond this simple explanation, the underlying mechanisms of the oxygen ion conductivity were not known.
In my doctoral thesis, I made the striking discovery that the ionic conductivity maximum is easily understood by separating the energy barriers for oxygen ion jumps around dopant ions in symmetric and asymmetric contributions. If the presence of a dopant changes the migration energy identically for forward and backward jumps, the resulting energy contribution is referred to as blocking. If the change in migration energy due to doping is different for a forward and backward jump of a specific ionic configuration, the resulting energy contributions are referred to as trapping. I have analyzed the influence of both effects on the ionic conductivity: Blocking determines the dopant fraction where the ionic conductivity exhibits the maximum. Trapping limits the maximum ionic conductivity value. I have found that trapping is a two‑step process (catch‑and‑hold) and that the lowest level of trapping for both steps results in the material with the highest ionic conductivity. In this way, the material leading to the highest conductivity can be predicted using only a few calculated energy barriers and new materials for catalysis and energy conversion can be developed.
Indeed, ionic conductivity maxima are found in doped oxides with the fluorite structure (ceria, zirconia, hafnia), perovskite structure (gallates, aluminates), and apatite structure, without the creation of oxygen vacancies (bismuth oxide) and even mixed ionic-electronic conductors (MIEC) such as BSCF, proton conductivity in barium zirconate or Li‑conducting NASICON materials. Therefore, the detailed results and insights obtained here for doped ceria may be generalized and applied to other ion conductors that are important for SOFCs, SOECs, and resistive switching materials, as well as solid-state batteries.
I have reviewed and classified over 1000 publications and performed impedance spectroscopy experiments, DFT calculations using the Vienna Ab initio Simulation Package (VASP), and KMC simulations concerning the ionic conductivity. The calculations agree with the experiments. I have published the results in a high-profile Perspective article in PCCP. Moreover, my article and cover art were showcased through being chosen as the PCCP outside front cover.
![]() MD
DFT
metadynamics
TST
phonon calculations
MD
DFT
metadynamics
TST
phonon calculations
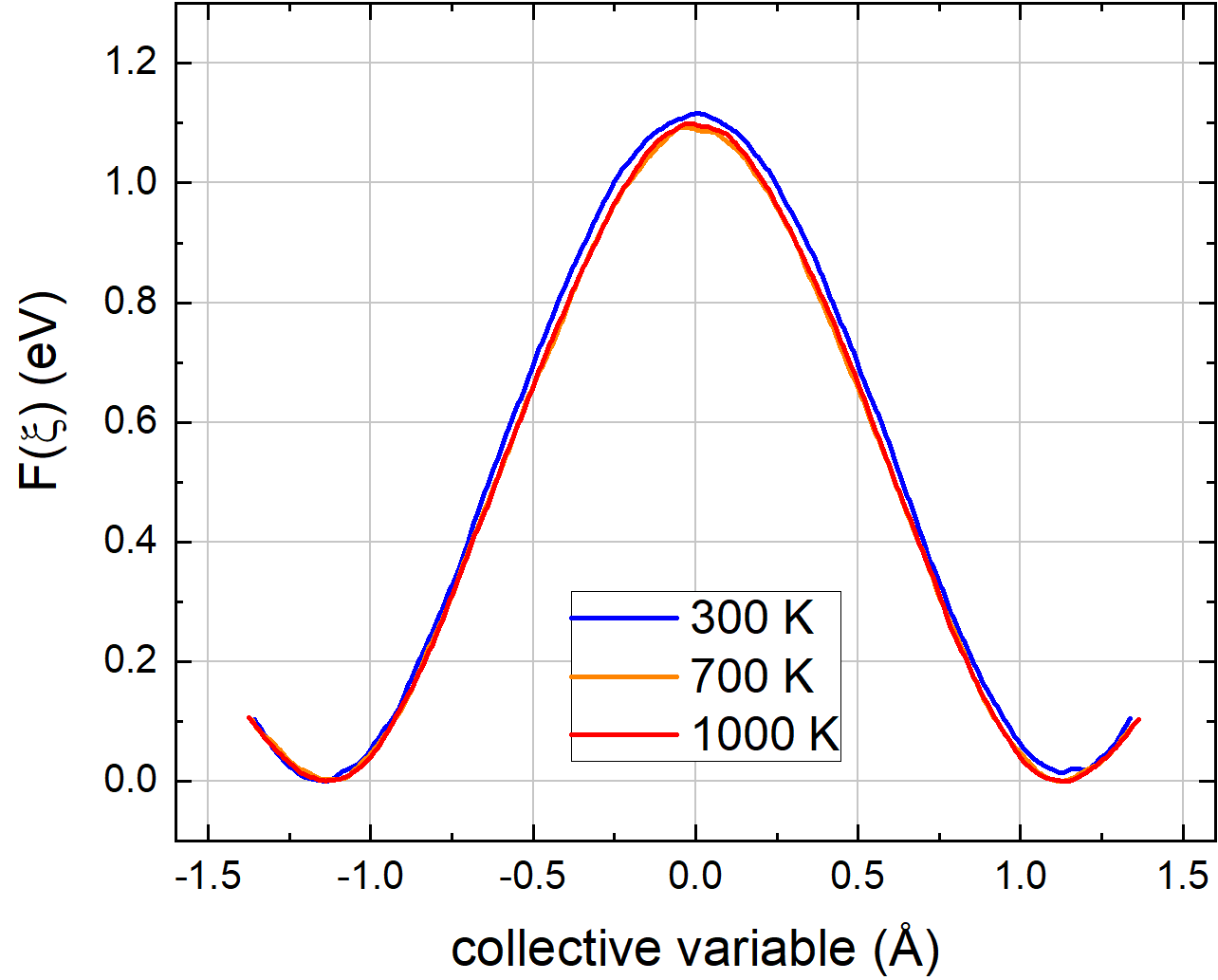
Computer simulations of activated processes, which play an important role in chemistry and materials physics, have, until recently, been limited to the static approach based on atomic relaxations. As a result, only the electronic energy of activation is calculated while, for vibrational and entropic contributions, a typical value is assumed. In my ![]() PCCP article (2017), I calculated the attempt frequency of oxygen ion jumps from first principles in order to allow a fully ab initio prediction of the ionic conductivity using phonon calculations of the oxygen ion migration. My article provides an in-depth overview of the transition state theory (TST) and shows the equivalence of the Eyring and Vineyard method without restriction to the Gamma point. Following this, I decided to change my topic. In my
PCCP article (2017), I calculated the attempt frequency of oxygen ion jumps from first principles in order to allow a fully ab initio prediction of the ionic conductivity using phonon calculations of the oxygen ion migration. My article provides an in-depth overview of the transition state theory (TST) and shows the equivalence of the Eyring and Vineyard method without restriction to the Gamma point. Following this, I decided to change my topic. In my ![]() Physical Review B article (2018), the restrictions of the harmonic approximation, such as the presumed shape of the potential energy surface and the decoupling of vibrational modes, are lifted and I take anharmonicity effects into account by using ab initio molecular dynamics (MD, DFT) combined with the metadynamics method. In metadynamics, a time-dependent bias potential is added to the Lagrangian driving molecular dynamics until the migration barrier no longer exists and the bias potential represents a negative image of the free energy profile. In other words, by filling up the double-well free energy potential for the oxygen ion jump, we see the activation energy and entropy of the migration barrier. In my articles, I discuss both the influence of a thermally expanding cell volume on the harmonic frequencies as well as the difference between Helmholtz and Gibbs free energies of activation.
Physical Review B article (2018), the restrictions of the harmonic approximation, such as the presumed shape of the potential energy surface and the decoupling of vibrational modes, are lifted and I take anharmonicity effects into account by using ab initio molecular dynamics (MD, DFT) combined with the metadynamics method. In metadynamics, a time-dependent bias potential is added to the Lagrangian driving molecular dynamics until the migration barrier no longer exists and the bias potential represents a negative image of the free energy profile. In other words, by filling up the double-well free energy potential for the oxygen ion jump, we see the activation energy and entropy of the migration barrier. In my articles, I discuss both the influence of a thermally expanding cell volume on the harmonic frequencies as well as the difference between Helmholtz and Gibbs free energies of activation.
![]() DFT
MMC
KMC
XANES
EXAFS
impedance spectroscopy
DFT
MMC
KMC
XANES
EXAFS
impedance spectroscopy
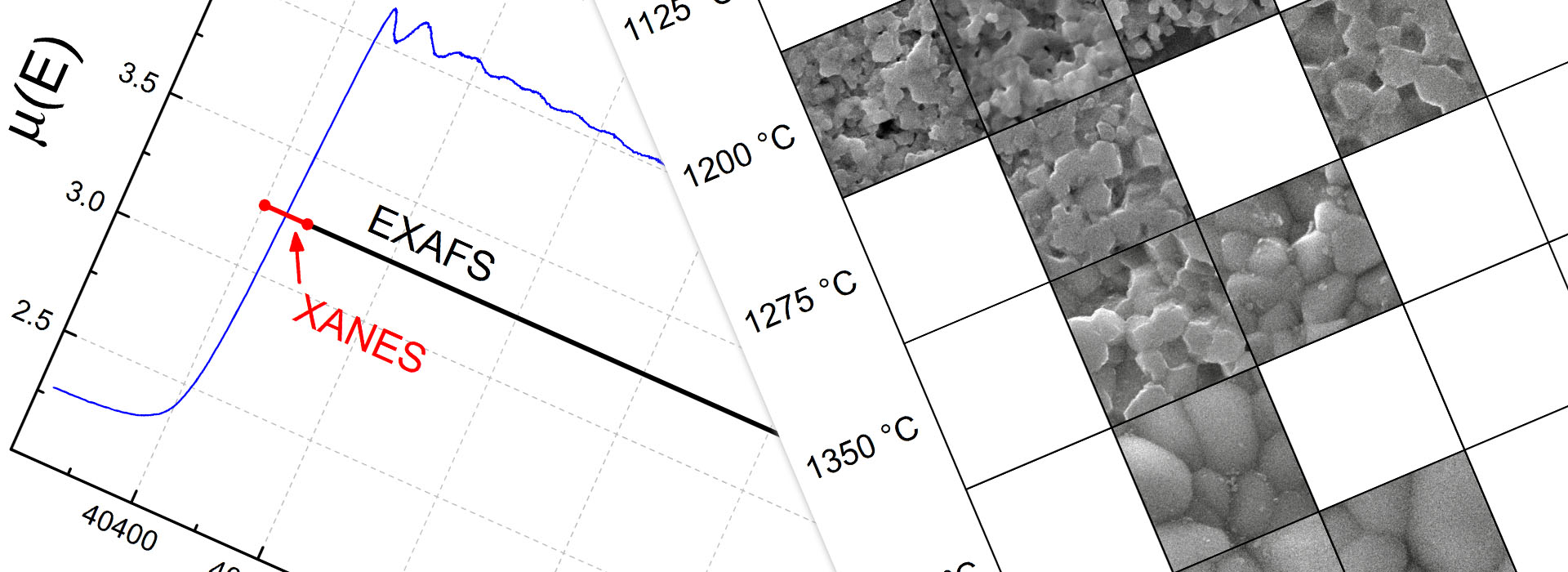
In doped ceria, dopant ions and oxygen vacancies interact. Both an association of defects, e.g. between dopants and oxygen vacancies, and a repulsion of defects, e.g. between oxygen vacancies, are found. I calculated defect interactions and association energies using DFT calculations. Here, I have accounted for the localization of the strongly correlated f‑electrons of cerium by introducing a Hubbard U parameter while investigating charge localization and electronic structure. Combined with Metropolis Monte Carlo (MMC) simulation, the association energies are employed to simulate the local structure. The resulting coordination numbers are in agreement with X‑ray absorption spectroscopy (XAS) measurements, which I performed at the German Electron Synchrotron (DESY). While I investigated for the X‑ray absorption near-edge structure (XANES), the valence state for the cerium cations in doped ceria, extended X‑ray absorption fine structure (EXAFS) measurements provided the coordination number of cations and anions. I used both the amplitude of the radial distribution function from EXAFS measurements as well as the distance to the neighboring atoms. For the latter, I improved the accuracy of the coordination numbers from EXAFS experiments. In our PCCP article (2014), we found a long-term degradation of the ionic conductivity due to cation ordering, which I also found in impedance spectroscopy measurements in my doctoral thesis.
![]() impedance spectroscopy
XRD
EDX
SEM
DFT
KMC
impedance spectroscopy
XRD
EDX
SEM
DFT
KMC
Samples of Sm and Lu doped ceria (Ce1‑xRExO2‑x/2 with 0 < x < 0.25 in steps of Δx = 0.025) were synthesized using the sol-gel method, pressed to pellets and characterized using X‑ray diffraction (XRD), energy dispersive X‑ray spectroscopy (EDX), density measurements and scanning electron microscopy (SEM). The influence of the sintering temperature and duration on the density, grain size and ionic conductivity using impedance spectroscopy measurements of the bulk and grain boundary domain was shown. The results for the polycrystalline samples were compared with single crystal samples and co-doped ceria. Finally, the effect of small amounts of impurity was investigated. The experiments are in agreement with Kinetic Monte Carlo (KMC) simulations based on density functional theory (DFT) calculations.
2012 – 2020
Applications for high-performance computing resources:
2013 – 2019
16 Progress reports and 6 annual reports for high-performance computing resources